A lightweight bridge from molecular structures to descriptors, contact maps, graph-ML inputs, and educational coarse-grained representations.
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers

Project description

MolScope








Turn a molecular structure file into descriptors, contact maps, ML graphs, and coarse-grained bead models, with a small, readable Python API.
Reads .xyz, .pdb, .cif, and .sdf (or fetches from the RCSB by ID). The
core depends only on NumPy and Matplotlib; heavier backends (RDKit, PyTorch
Geometric, DGL, Gemmi) are opt-in extras. Built for teaching, exploratory
analysis, and ML-for-molecules prototyping, not as a replacement for full
simulation or cheminformatics stacks.
📖 Full documentation: https://molscope.readthedocs.io
| 3D structure (element) | Secondary structure (DSSP) | Residue contact map | Coarse-grained beads |
|---|---|---|---|
 |
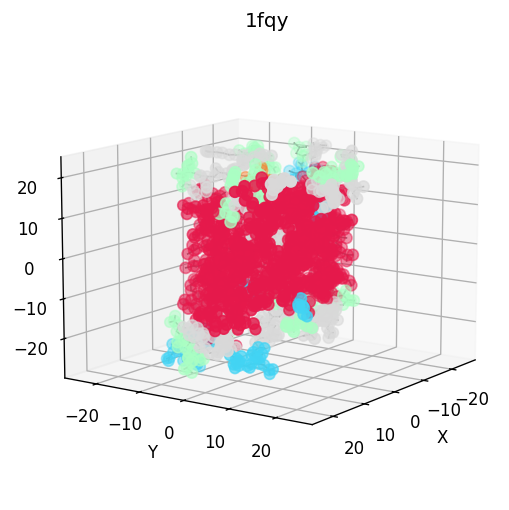 |
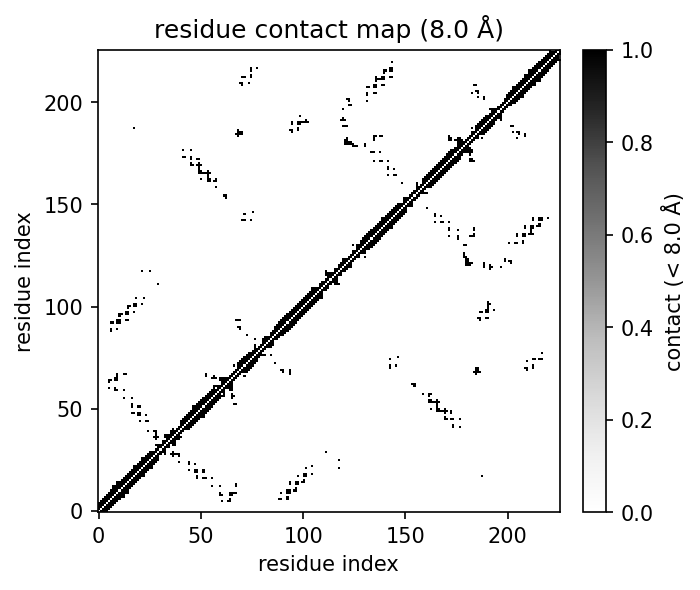 |
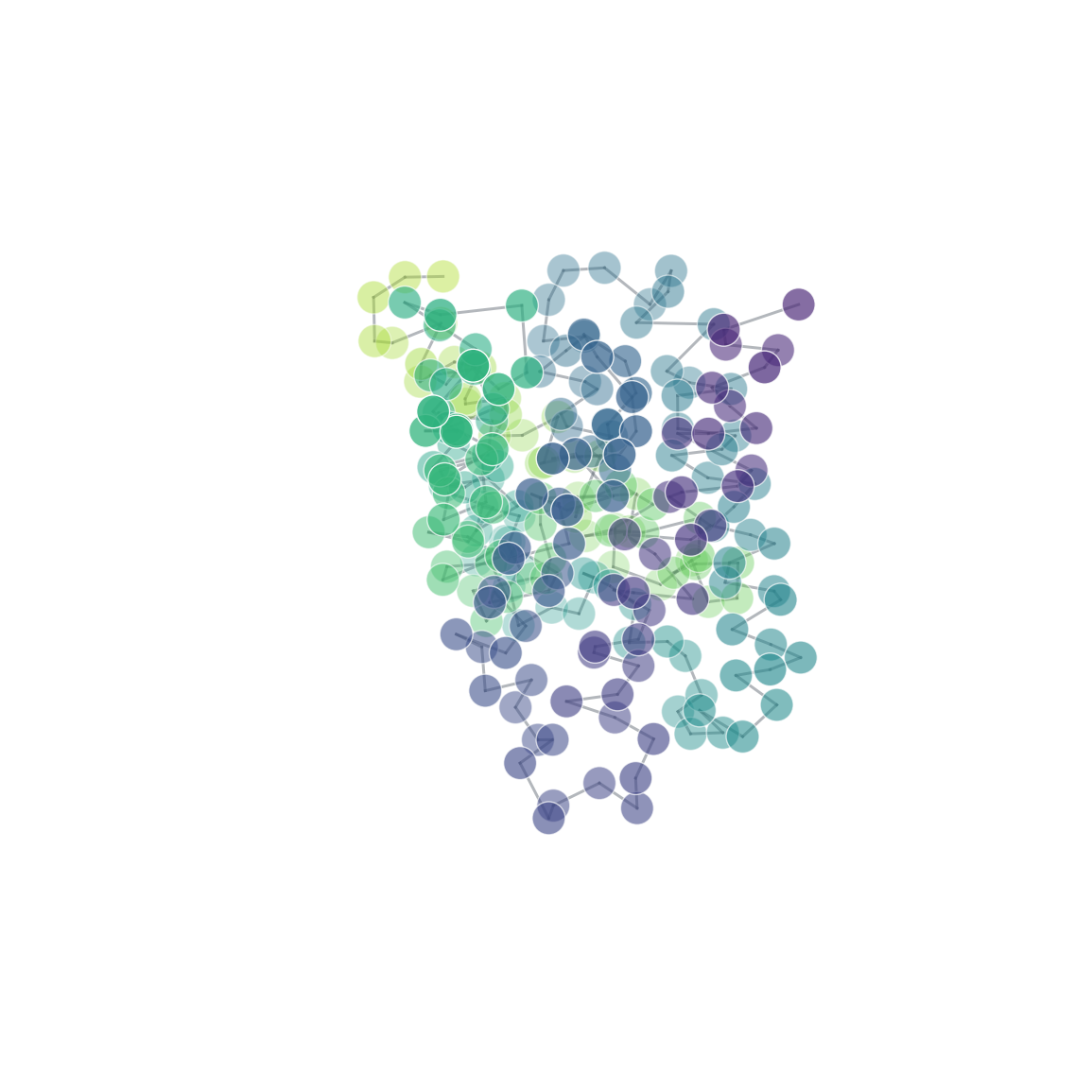 |
Install
pip install molscope # core: NumPy + Matplotlib only
Optional extras, added only when a workflow needs them:
| Extra | Adds |
|---|---|
fast |
scipy KD-tree for faster bond/contact search on large structures |
chem |
RDKit chemical perception and descriptors |
cif |
Gemmi mmCIF parsing and validation |
pyg / dgl / graph / gnn |
PyTorch Geometric / DGL / NetworkX graph export |
viz |
py3Dmol interactive viewer |
xlsx |
read/write .xlsx molecule tables |
gpu |
Torch dense distance backend |
mcp |
MCP server for AI assistants (Python >= 3.10) |
pip install "molscope[chem,cif,pyg]" # combine as needed
For local development with uv: uv sync (creates
.venv and installs deps + dev tools from the lockfile), then uv run pytest.
Quickstart
Given a .pdb (or .xyz / .cif / .sdf), here is what you can pull out:
import molscope as ms
mol = ms.read("protein.pdb") # or ms.fetch("1fqy") from the RCSB
print(mol.summary()) # atoms, formula, chains, bounding box
ca = mol.select(chain="A").alpha_carbons() # metadata selections
cmap = mol.contact_map(cutoff=8.0) # residue contact map (NumPy)
desc = mol.descriptors() # dict of structural descriptors
graph = mol.to_graph() # ML-ready graph, no extra deps
data = mol.to_pyg_data() # PyTorch Geometric Data ([pyg])
cg = mol.coarse_grain("residue_com") # one bead per residue
Molecule is immutable: translate, centered, and rotate each return a new
molecule, so transformations chain cleanly.
What you can do
| Capability | Guide |
|---|---|
| Read/write XYZ, PDB, mmCIF, SDF; fetch from RCSB; build from SMILES | Reading files |
| Stream large multi-model files frame by frame | Reading files |
| Select atoms by metadata (chain, residue, name, ...) | Selections |
| Geometry, RMSD, distances, angles, torsions | Geometry and measurements |
| Contact maps and distance matrices | Contact maps |
| DSSP secondary structure, torsions, interfaces, binding sites | Protein analysis |
| Native and RDKit-backed descriptors | Structural descriptors |
| Chemical perception, protein template bonds, bond-order inference | Chemical perception |
| Atom/bond and residue-contact graphs for ML (with positional encodings) | Molecular graphs |
| Coarse-grained bead mappings (residue, Martini-style, custom) | Coarse-graining |
| NMR ensembles and clustering | Ensemble analysis |
| Plotting and py3Dmol viewing | Plotting and viewing |
| Diverse subset selection from a CSV/XLSX table | Diverse selection |
Task-oriented tutorials: PDB to descriptors,
PDB to graph/GNN, and
PDB to coarse-grained beads. A
runnable tour over the bundled samples lives in examples/tour.py.
Command line
| Command | Does |
|---|---|
molscope <file> (view) |
visualise a structure, save a PNG or GIF |
molscope analyze |
batch descriptor table to CSV |
molscope binding-site |
ligand binding-site contacts and pocket descriptors |
molscope export |
batch graph export to PyG / DGL / NetworkX |
molscope coarse-grain |
map a structure to CG beads and write a coordinate file (PDB CONECT bonds) |
molscope select |
diverse subset from a CSV/XLSX table |
molscope dock-summary |
rank docking poses from an SDF; summary + top-hit tables + score plot |
molscope dock-diverse |
diverse shortlist of top hits by Tanimoto clustering |
molscope dock-rank |
transparent consensus ranking across scored SDFs |
molscope dock-report |
self-contained HTML report + top poses for PyMOL/ChimeraX/Mol* |
molscope examples/data/1fqy.pdb --select atom_name=CA --color-by residue --save ca.png
molscope analyze examples/data/*.pdb --out results.csv --preset native-3d --jobs 4
molscope export "data/*.cif" --to pyg --out-dir pyg_graphs/ --pe laplacian --jobs 8
molscope coarse-grain examples/data/1fqy.pdb --mapping martini --out cg.pdb
molscope select molecules.csv --smiles-col SMILES --compute-descriptors -n 100 --out picked.csv
molscope dock-summary vina_out.sdf --score-field minimizedAffinity --top 20
molscope dock-diverse vina_out.sdf --top 500 --select 50
Use from an AI assistant (MCP)
MolScope ships an optional Model Context Protocol server, so an MCP-capable assistant (Claude Code/Desktop, Codex CLI, Gemini CLI) can drive its analyses in natural language. It exposes the public API as 28 tools (structure analysis, graphs, plots, dataset prep, docking-hit triage) and adds no new science.
pip install "molscope[mcp]" # needs Python >= 3.10
claude mcp add molscope -- molscope-mcp # Claude Code
codex mcp add molscope -- molscope-mcp # Codex CLI
gemini mcp add molscope molscope-mcp # Gemini CLI
For example: "fetch trypsin (3ptb), find the benzamidine binding-site residues,
and render a contact map." See docs/user-guide/mcp-server.md
for the full tool reference.
Scientific validation
MolScope is explicit about which results are cross-checked against reference tools and which are intentionally lightweight:
| Feature | Status |
|---|---|
| Geometry, RMSD, contact maps | Cross-checked vs MDAnalysis (near machine precision) |
| Bond perception, chemical features | Cross-checked vs RDKit |
| Secondary structure (simplified DSSP) | Cross-checked vs mkdssp: ~98 to 99% 3-state agreement across helical, mixed, and all-beta folds |
| Protein template bonds | Cross-checked vs known per-residue chemistry |
| Native descriptors, molecular graphs | Deterministic; not benchmarked against a curated library |
| Coarse-graining | Mapping and visualisation only; not a validated force-field model |
| Standard protonation | Idealised pH-7 textbook model ("standard") |
| pKa-aware protonation | Environment-aware via PROPKA (proteins) / Dimorphite-DL (SMILES) at a chosen pH |
Methods, tolerances, and failure modes are in docs/validation.md.
The CI validation job runs physical invariants plus these cross-checks on every push.
Scope and design philosophy
MolScope is a lightweight core (NumPy and Matplotlib only) with a broad optional surface. Those optional extras are deliberately of two kinds, and the distinction is what keeps "lightweight" honest:
- Interop / output targets (
networkx,pyg,dgl,viz,xlsx,gpu,mcp). These let MolScope emit to your framework or run faster. Speaking many formats is the bridge mission, so this surface is expected to grow. - Method backends (
chem/RDKit,cif/gemmi,propka,dimorphite,validation/MDAnalysis). These delegate a scientific computation to an external tool, and each one inherits that tool's scope, versioning, and correctness surface. This is the surface we keep deliberately small.
A new method backend earns inclusion only when MolScope adds real integration value beyond what you could write in a line or two against the tool directly (for example, mapping RDKit perception back onto MolScope's atom model and residue templates), and when the wrapped tool is maintained. The core does something useful on its own; everything heavier degrades gracefully when its extra is absent. MolScope is not, and is not trying to become, a re-implementation of a cheminformatics or simulation stack: where a dedicated tool is the right answer, it integrates that tool rather than reinventing it, and says so.
FAQ
Which formats can it read? .xyz, .pdb, .cif, and .sdf; fetch from the
RCSB with ms.fetch("1fqy"); or build from SMILES with ms.read_smiles(...)
(needs [chem]).
Does it handle MD trajectories? It works on static structures and multi-model
files (NMR ensembles, and ms.stream(...) to iterate large multi-model PDB/XYZ
frame by frame). It has no trajectory engine; for DCD/XTC and friends use
MDAnalysis or MDTraj.
Is the coarse-graining a real force field? No. It produces CG mappings and bead graphs for inspection and ML prototyping. The OpenMM XML export describes topology only and is not a validated Martini parameter set.
Do I need RDKit or PyTorch? No. The core runs on NumPy and Matplotlib; those are opt-in extras you install only for the matching workflow.
Will odd PDB files parse? ATOM/HETATM lines are read by fixed columns (not whitespace), so touching, large, or negative coordinate fields read correctly. Alternate conformations default to the primary altLoc.
Development and citation
uv run pytest # full test suite
uv run pytest tests/validation # validation suite only
uv run ruff check . # lint
CI runs the suite and linting across Python 3.9 / 3.11 / 3.13, smoke-imports the
extras, and runs a separate validation job on every push and PR. Notable changes
per release are in CHANGELOG.md.
Each release is archived on Zenodo with a citable DOI. The concept DOI
10.5281/zenodo.20433850 always resolves
to the latest version; citation metadata is in CITATION.cff, so
GitHub's "Cite this repository" button produces BibTeX and APA entries.
License
Project details
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file molscope-0.13.0.tar.gz.
File metadata
- Download URL: molscope-0.13.0.tar.gz
- Upload date:
- Size: 3.1 MB
- Tags: Source
- Uploaded using Trusted Publishing? Yes
- Uploaded via: uv/0.11.18 {"installer":{"name":"uv","version":"0.11.18","subcommand":["publish"]},"python":null,"implementation":{"name":null,"version":null},"distro":{"name":"Ubuntu","version":"24.04","id":"noble","libc":null},"system":{"name":null,"release":null},"cpu":null,"openssl_version":null,"setuptools_version":null,"rustc_version":null,"ci":true}
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
84190fa87b6671c87c91f71466850e56dfa785d3de693724d77c46c7f566a6fb
|
|
| MD5 |
a19bd8b4a504de7293836f58d4d19cab
|
|
| BLAKE2b-256 |
2bd0e1fd88280bfc91cc63febf885501a1aa141e32460179078f951acc140183
|
File details
Details for the file molscope-0.13.0-py3-none-any.whl.
File metadata
- Download URL: molscope-0.13.0-py3-none-any.whl
- Upload date:
- Size: 177.1 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? Yes
- Uploaded via: uv/0.11.18 {"installer":{"name":"uv","version":"0.11.18","subcommand":["publish"]},"python":null,"implementation":{"name":null,"version":null},"distro":{"name":"Ubuntu","version":"24.04","id":"noble","libc":null},"system":{"name":null,"release":null},"cpu":null,"openssl_version":null,"setuptools_version":null,"rustc_version":null,"ci":true}
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
5a5ea9bfd12a0b2cb40689b30ef90938ec2e9d572825d68f509c66b6d9891209
|
|
| MD5 |
b4e4b993032c7ea43cc3b0b3527ecfff
|
|
| BLAKE2b-256 |
003a31b6200c91ea7a89f0011e9c20997db592e777ca052f6767bbc12087e3a3
|







