Lightweight molecular structure analysis, visualisation, graph export, and coarse-graining in Python.
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers

Project description
MolScope




Lightweight molecular structure analysis, visualisation, graph export, and
coarse-graining in Python. Read .xyz, .pdb, .cif and .sdf files
(optionally gzip-compressed), select and analyse atoms, and visualise them in
3D. The .cif reader is a basic mmCIF parser for standard _atom_site
coordinate loops, not a full mmCIF syntax implementation.
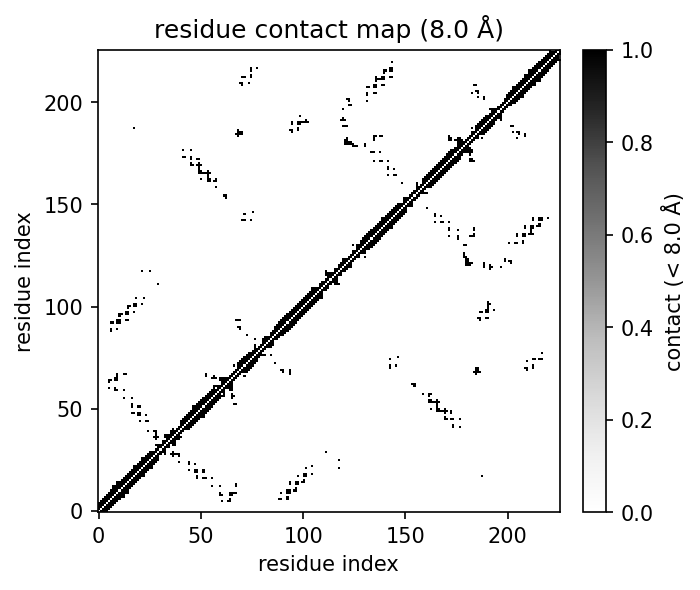
| 3D structure rendering | Residue contact map | Coarse-grained beads |
|---|---|---|
 |
 |
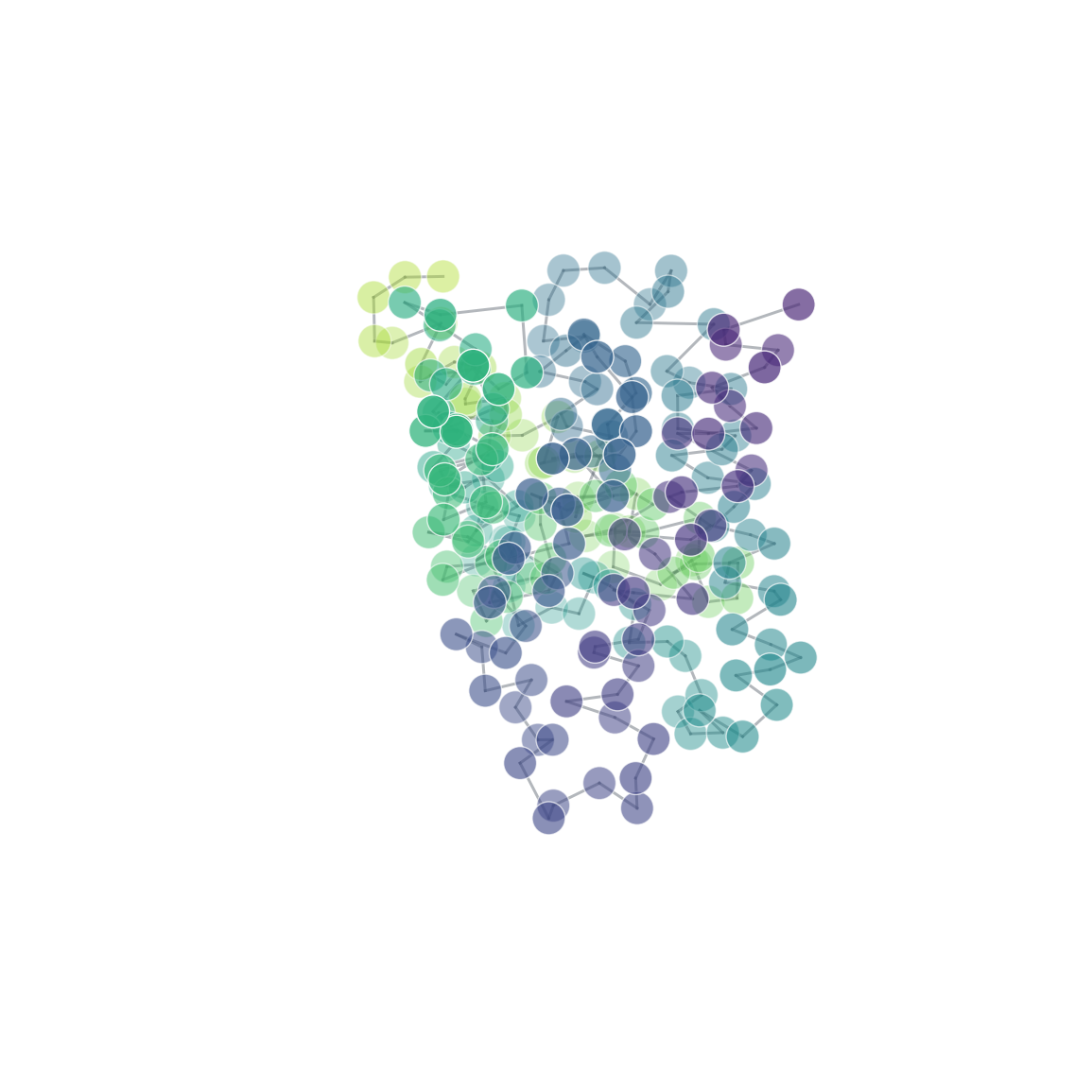 |
What it does
- Read and write XYZ, PDB, mmCIF and SDF (gzip-aware), fetch structures by id from RCSB, and load multi-model NMR ensembles.
- Select and measure by chain, element or residue; compute distances, angles, dihedrals and Kabsch-aligned RMSD.
- Analyse centroids, radius of gyration, the inertia tensor, inferred bonds and contacts.
- Contact maps at atom or residue level, with heatmap plots.
- Ensembles: pairwise RMSD, RMSF, averaging, and conformer clustering.
- Export for ML: flat structural descriptors and molecular graphs for NetworkX, PyTorch Geometric and DGL.
- Coarse-grain onto residue, Martini-style or custom bead mappings.
- Visualise with 3D matplotlib plots, an interactive py3Dmol viewer, spin GIFs, and a command-line interface.
Install
With uv (recommended):
uv sync # creates .venv, installs deps + dev tools from the lockfile
uv run molscope 1fqy.pdb # run the CLI
uv run pytest # run the tests
uv sync pins the interpreter from .python-version and resolves against
uv.lock for reproducible installs. Use uv sync --no-dev to skip the test tools.
With plain pip:
python -m venv .venv && source .venv/bin/activate
pip install -e ".[test]" # or: pip install -r requirements.txt
Documentation
The documentation website is built with MkDocs Material:
uv sync --group docs
uv run mkdocs serve
python scripts/build_user_guide_pdf.py
Docs source lives in docs/; the site configuration is mkdocs.yml. The PDF
builder requires Pandoc and a LaTeX engine such as xelatex.
Quickstart
A runnable end-to-end tour over the bundled sample structures lives in
example.py:
uv run python example.py # opens 3D plot windows
MPLBACKEND=Agg uv run python example.py # headless: saves PNGs instead
It reads an .xyz and a .pdb, prints derived properties, compares the NMR
models of 1aml, writes a transformed structure back out, and renders a plot.
Library
import molscope as ms
mol = ms.read("1fqy.pdb") # parser chosen from the extension
mol = ms.fetch("1fqy") # ...or download straight from RCSB by id
print(mol.summary()) # atoms, formula, chains, bounding box
mol = mol.centered().rotate("z", 90).translate((1, 2, -1))
mol.plot() # CPK colours, inferred bonds, equal aspect
Molecule is immutable: translate, centered and rotate each return a new
molecule, so transformations chain cleanly without aliasing. Equality is by
value (np.array_equal on coordinates).
Selections
PDB files, and standard mmCIF atom-site loops, carry per-atom metadata (atom name, residue, chain), so you can slice a structure:
mol.select(chain="A") # one chain
mol.select(element="C") # all carbons
mol.select(resname="HOH") # waters
mol.select(resid=(10, 20)) # an inclusive residue range
mol.alpha_carbons() # CA atoms (the usual basis for protein RMSD)
mol.backbone() # N, CA, C, O
mol[mask_or_indices] # subset by numpy mask / index array
Analysis and measurements
mol.centroid, mol.center_of_mass # geometric / mass-weighted centre
mol.radius_of_gyration # compactness (angstrom)
mol.dimensions, mol.formula # bounding box, Hill-order formula
mol.bonds() # inferred bond index pairs (KD-tree if scipy)
mol.contacts(cutoff=5.0) # atom pairs within a distance
mol.distance(i, j) # bond length
mol.angle(i, j, k) # bond angle (degrees)
mol.dihedral(a, b, c, d) # torsion angle (degrees)
a.alpha_carbons().rmsd(b.alpha_carbons(), align=True) # CA-RMSD after Kabsch fit
Structural descriptors for ML
features = mol.descriptors() # flat dict of scalar/vector descriptors
features["radius_of_gyration"]
features["principal_moments"] # 3 values
features["distance_histogram"] # fixed-size histogram
X, names = ms.featurize_many(
["a.pdb", "b.pdb", "c.xyz"],
return_names=True,
) # numeric matrix + column names
Descriptors include atom/residue counts, element counts, molecular mass,
centres, radius of gyration, bounding-box dimensions, inertia tensor, principal
moments/axes, shape anisotropy, compactness, distance histograms, bond-length
summary statistics, and atom/residue contact summaries. Full contact maps remain
available through mol.contact_map(...).
Contact maps
cmap = mol.contact_map(cutoff=8.0, level="residue") # CA-CA contacts -> ContactMap
cmap.matrix # (R, R) array
mol.plot_contact_map(cutoff=8.0) # heatmap
mol.contact_map(level="atom") # atom-level map
mol.contact_map(level="residue", method="min") # closest inter-residue atom
mol.contact_map(level="residue", method="com") # residue centre of mass
NMR ensembles
from molscope import ensemble
models = ms.read_pdb_models("1aml.pdb") # all 20 models
ensemble.rmsd_matrix(models) # pairwise RMSD matrix
ensemble.rmsf(models) # per-atom fluctuation
ensemble.average(models) # mean structure
ensemble.align_all(models) # superpose every model onto the first
# Per-residue-pair contact probability across the ensemble (NMR variability)
freq = ms.ensemble_contact_frequency(models, cutoff=8.0)
freq.plot() # heatmap of contact frequencies in [0, 1]
Comparing and clustering conformers
Cluster an ensemble (NMR models, conformer sets, docking poses, MD snapshots) by pairwise RMSD:
matrix = ms.rmsd_matrix(models, align=True) # (M, M) RMSD matrix
ms.plot_rmsd_heatmap(matrix) # heatmap
clusters = ms.cluster(models, method="hierarchical") # data-driven cutoff
clusters = ms.cluster(models, n_clusters=3) # ...or a fixed count
clusters.n_clusters # how many clusters
clusters.groups() # {cluster_id: [model indices]}
clusters.representatives() # {cluster_id: medoid model index}
ms.plot_rmsd_heatmap(matrix, order=clusters.order) # reorder into diagonal blocks
Writing and viewing
ms.write_xyz(mol.centered(), "out.xyz") # write transformed coordinates back
ms.write_pdb(mol, "out.pdb")
mol.plot(color_by="chain") # colour by element / chain / residue
mol.view(style="cartoon") # interactive py3Dmol viewer (notebooks)
from molscope.plotting import spin_gif
spin_gif(mol, "spin.gif") # rotating animation
Molecular graphs (for machine learning)
Turn 3D coordinates plus inferred bonds into a graph, then export to the common
ML frameworks. The base to_graph() needs no extra dependencies; each exporter
imports its backend lazily.
mol = ms.read("1fqy.pdb")
g = mol.to_graph() # MolecularGraph: nodes + edges, no deps
g.n_atoms, g.n_bonds # counts
g.atomic_numbers, g.masses # per-node arrays
g.node_features() # (N, 2) default features [atomic_number, mass]
G = mol.to_networkx() # networkx.Graph with node/edge attributes
data = mol.to_pyg_data() # torch_geometric.data.Data (x, pos, edge_index, edge_attr, z)
dglg = mol.to_dgl_graph() # dgl.DGLGraph with ndata/edata tensors
Nodes carry element, atomic number, mass, coordinates and (from PDB/mmCIF) atom
name, residue and chain. Edges carry the bonded pair, interatomic distance, and
bond order (1.0 for geometrically inferred bonds). Install backends as needed:
pip install "molscope[graph]" installs only NetworkX. PyTorch Geometric and
DGL are optional manual installs: pip install torch torch_geometric or
pip install dgl after choosing the right PyTorch build for your platform.
Coarse-graining
Map an atomistic structure onto a smaller set of beads. The result is an
ordinary Molecule (beads as "atoms") with explicit CG bonds attached, so it
plots, transforms and graphs like anything else.
mol = ms.read("1fqy.pdb")
cg = mol.coarse_grain("residue_com") # one bead per residue (centre of mass)
cg = mol.coarse_grain("residue_centroid") # ...or geometric centroid
cg = mol.coarse_grain("martini") # simplified backbone + side-chain beads
cg.plot(scale=200) # beads + backbone topology
print(cg.mapping_report()) # explain beads, dropped atoms, and bonds
# Custom bead definitions by residue + atom name (needs PDB/mmCIF metadata)
mapping = {"ALA": {"BB": ["N", "CA", "C", "O"], "SC": ["CB"]}}
cg = mol.coarse_grain(mapping)
cg, report = mol.coarse_grain(mapping, return_report=True)
# Custom bead definitions by atom index (works on ANY structure, even .xyz)
cg = mol.coarse_grain({"head": [0, 1, 2, 3], "tail": [4, 5, 6, 7]},
bonds=[("head", "tail")]) # define the bead network too
cg.to_graph() # CG bead network, ready for ML
Bead positions are mass-weighted (or centroids). For residue mappings bonds are
generated automatically (within a residue, plus a backbone chain between
residues); pass bonds= to define them yourself. Name-based bonds are intended
for unique bead names such as head/tail; repeated names such as BB/SC
are ambiguous, so use bead indices for those. Atoms you leave unassigned are
dropped with a warning. This is meant
for teaching and prototyping CG mappings, not as a replacement for production
Martini parameters.
Command line
molscope helix_201.xyz --translate 1 2 -1
molscope 1fqy.pdb --select atom_name=CA --color-by residue --save ca.png
molscope --fetch 1aml --center --gif amyloid.gif
python -m molscope 1fqy.pdb # equivalent if not pip-installed
Sample structures
| File | Contents |
|---|---|
helix_201.xyz |
a helix (bare coordinates) |
1fqy.pdb |
Aquaporin-1, single model (1661 atoms) |
1aml.pdb |
Alzheimer amyloid A4 peptide, 20-model NMR ensemble |
Notes
- PDB files are parsed by fixed columns, not whitespace splitting, so atoms with touching coordinate fields (large or negative values) read correctly.
- Alternate conformations (altLoc) other than the primary one are skipped.
read_pdbreturns a single model (model=1by default); useread_pdb_modelsfor the whole ensemble.- Bond inference uses a
scipy.spatial.cKDTreewhen available; without scipy it falls back to a denseO(n^2)search that is refused above ~8000 atoms. - Optional extras:
pip install "molscope[fast]"(scipy, faster bonds/contacts) and"molscope[viz]"(py3Dmol, forMolecule.view).
Tests and linting
uv run pytest # full test suite
uv run ruff check . # lint
CI (GitHub Actions) runs both across Python 3.9 / 3.11 / 3.13 on every push and PR.
License
Project details
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file molscope-0.6.2.tar.gz.
File metadata
- Download URL: molscope-0.6.2.tar.gz
- Upload date:
- Size: 47.1 kB
- Tags: Source
- Uploaded using Trusted Publishing? Yes
- Uploaded via: uv/0.11.16 {"installer":{"name":"uv","version":"0.11.16","subcommand":["publish"]},"python":null,"implementation":{"name":null,"version":null},"distro":{"name":"Ubuntu","version":"24.04","id":"noble","libc":null},"system":{"name":null,"release":null},"cpu":null,"openssl_version":null,"setuptools_version":null,"rustc_version":null,"ci":true}
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
f3f8c283d7003b3a42867819bdfea99dc8d05c55dddaafd3f1a32217ff91dfbe
|
|
| MD5 |
fb4ff6bf40331a0565b0cc28a0c36b4f
|
|
| BLAKE2b-256 |
78264ea0c2d5d6acb71b421cf43d60c9b10c8ec3ac8aae4b98d8e2346ea7bdce
|
File details
Details for the file molscope-0.6.2-py3-none-any.whl.
File metadata
- Download URL: molscope-0.6.2-py3-none-any.whl
- Upload date:
- Size: 38.4 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? Yes
- Uploaded via: uv/0.11.16 {"installer":{"name":"uv","version":"0.11.16","subcommand":["publish"]},"python":null,"implementation":{"name":null,"version":null},"distro":{"name":"Ubuntu","version":"24.04","id":"noble","libc":null},"system":{"name":null,"release":null},"cpu":null,"openssl_version":null,"setuptools_version":null,"rustc_version":null,"ci":true}
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
fb880bbdd492748194a65c58c7e0b2ca893d1b017c96b3f9ab98f30f2fc66e1e
|
|
| MD5 |
bf8ecb74b63dd0c6bc0b90078393cbfc
|
|
| BLAKE2b-256 |
344c30c6a9edac69b2911a212e27df0d790ea867e1bee9e97c8f561ad74aabc9
|







