MS2PIP: Accurate and versatile peptide fragmentation spectrum prediction.
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers



Project description









MS²PIP: MS2 Peak Intensity Prediction - Fast and accurate peptide fragmentation spectrum prediction for multiple fragmentation methods, instruments and labeling techniques.
About
MS²PIP is a tool to predict MS2 peak intensities from peptide sequences. The result is a predicted peptide fragmentation spectrum that accurately resembles its observed equivalent. These predictions can be used to validate peptide identifications, generate proteome-wide spectral libraries, or to select discriminative transitions for targeted proteomics. MS²PIP employs the XGBoost machine learning algorithm and is written in Python and C.
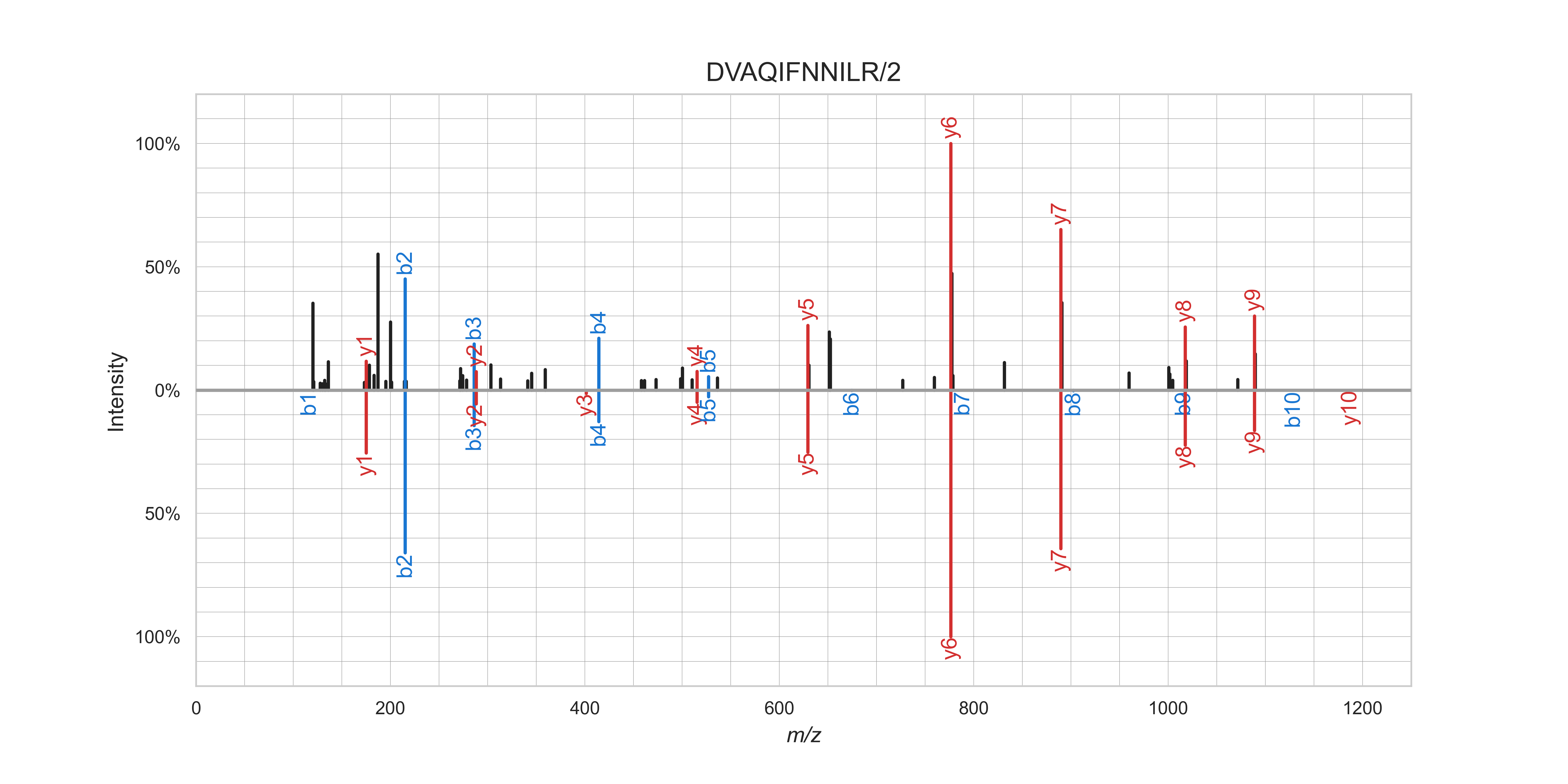
Mirror plot of an observed (top) and MS²PIP-predicted (bottom) spectrum for the peptide DVAQIFNNILR/2.
You can install MS²PIP on your machine by following the installation instructions. For a more user-friendly experience, go to the MS²PIP web server. There, you can easily upload a list of peptide sequences, after which the corresponding predicted MS2 spectra can be downloaded in multiple file formats. The web server can also be contacted through the RESTful API.
The MS³PIP Python application can perform the following tasks:
predict-single: Predict fragmentation spectrum for a single peptide and optionally visualize the spectrum.
predict-batch: Predict fragmentation spectra for a batch of peptides.
predict-library: Predict a spectral library from protein FASTA file.
correlate: Compare predicted and observed intensities and optionally compute correlations.
correlate-single: Compare predicted and observed intensities for a single peptide spectrum.
get-training-data: Extract feature vectors and target intensities from observed spectra for training.
annotate-spectra: Annotate peaks in observed spectra.
MS²PIP supports a wide range of PSM input formats and spectrum output formats, and includes pre-trained models for multiple fragmentation methods, instruments and labeling techniques. See Usage for more information.
Citations
If you use MS²PIP for your research, please cite the following publication:
Declercq, A., Bouwmeester, R., Chiva, C., Sabidó, E., Hirschler, A., Carapito, C., Martens, L., Degroeve, S., Gabriels, R. (2023). Updated MS²PIP web server supports cutting-edge proteomics applications. Nucleic Acids Research doi:10.1093/nar/gkad335
Prior MS²PIP publications:
Gabriels, R., Martens, L., & Degroeve, S. (2019). Updated MS²PIP web server delivers fast and accurate MS2 peak intensity prediction for multiple fragmentation methods, instruments and labeling techniques. Nucleic Acids Research doi:10.1093/nar/gkz299
Degroeve, S., Maddelein, D., & Martens, L. (2015). MS²PIP prediction server: compute and visualize MS2 peak intensity predictions for CID and HCD fragmentation. _Nucleic Acids Research, 43(W1), W326–W330. doi:10.1093/nar/gkv542
Degroeve, S., & Martens, L. (2013). MS²PIP: a tool for MS/MS peak intensity prediction. Bioinformatics (Oxford, England), 29(24), 3199–203. doi:10.1093/bioinformatics/btt544
Please also take note of, and mention, the MS²PIP version you used.
Full documentation
The full documentation, including installation instructions, usage examples, and the command-line and Python API reference, can be found at ms2pip.readthedocs.io.
Contributing
Bugs, questions or suggestions? Feel free to post an issue in the issue tracker or to make a pull request. Any contribution, small or large, is welcome!
Project details
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distributions
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file ms2pip-4.1.2.tar.gz.
File metadata
- Download URL: ms2pip-4.1.2.tar.gz
- Upload date:
- Size: 5.5 MB
- Tags: Source
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
beadaf4a703d99885ea49d7b2411752c2a4d7f5abf266878ed254c22b869b015
|
|
| MD5 |
1895bcd4368759776e4b8d92f6838a54
|
|
| BLAKE2b-256 |
f168c004586e697ebd4240afcbe8cae11ccf68de6737e9ef97dc672dfb1863c6
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2.tar.gz:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2.tar.gz -
Subject digest:
beadaf4a703d99885ea49d7b2411752c2a4d7f5abf266878ed254c22b869b015 - Sigstore transparency entry: 937226501
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp313-cp313-win_amd64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp313-cp313-win_amd64.whl
- Upload date:
- Size: 13.6 MB
- Tags: CPython 3.13, Windows x86-64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
fb4d72b15ca6533bd83a5fc028c62148316854580a4321131579dbac8dc337b2
|
|
| MD5 |
2f85fac1218f96ceeb08ecb801129b5e
|
|
| BLAKE2b-256 |
f0cf241110bc5d715e21bfb955c37ec1a1d80a3e688e581d4bdd644791e6a8d1
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp313-cp313-win_amd64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp313-cp313-win_amd64.whl -
Subject digest:
fb4d72b15ca6533bd83a5fc028c62148316854580a4321131579dbac8dc337b2 - Sigstore transparency entry: 937226571
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp313-cp313-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp313-cp313-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl
- Upload date:
- Size: 15.5 MB
- Tags: CPython 3.13, manylinux: glibc 2.17+ x86-64, manylinux: glibc 2.5+ x86-64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
9d66b97ac898ffd7c10b2babf64956d86f1f497533504c8157650353346dfb06
|
|
| MD5 |
65be70630aa8748b53a447e65eeceed2
|
|
| BLAKE2b-256 |
40f70b90d0cef896ab30f5be8d3cf8321f3ec3559af5bc44ab95de72a3b95de8
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp313-cp313-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp313-cp313-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl -
Subject digest:
9d66b97ac898ffd7c10b2babf64956d86f1f497533504c8157650353346dfb06 - Sigstore transparency entry: 937226711
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp313-cp313-macosx_11_0_arm64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp313-cp313-macosx_11_0_arm64.whl
- Upload date:
- Size: 14.8 MB
- Tags: CPython 3.13, macOS 11.0+ ARM64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
f16ae2764a6017eb6c24759cc721ee7f94ef822563bb69e356dda49d08bc01fb
|
|
| MD5 |
31869993969e6cf544ab101cb47d9bef
|
|
| BLAKE2b-256 |
1bbc40ec44f714ebd9d0bdb2421cf44c1f294aad7ef0dcfc7644763594929b58
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp313-cp313-macosx_11_0_arm64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp313-cp313-macosx_11_0_arm64.whl -
Subject digest:
f16ae2764a6017eb6c24759cc721ee7f94ef822563bb69e356dda49d08bc01fb - Sigstore transparency entry: 937226643
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp313-cp313-macosx_10_13_x86_64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp313-cp313-macosx_10_13_x86_64.whl
- Upload date:
- Size: 13.9 MB
- Tags: CPython 3.13, macOS 10.13+ x86-64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
9cb9c44f8130b7e1144fef4554fd972b9df28d70bdfee2b20f5c7c32c1ab7274
|
|
| MD5 |
8006990fc8f65822c7f48b597ca966b9
|
|
| BLAKE2b-256 |
4db4c04b2e69addd0231f1620ac6376dfb7d12e38117212be9787b2b9f3252c3
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp313-cp313-macosx_10_13_x86_64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp313-cp313-macosx_10_13_x86_64.whl -
Subject digest:
9cb9c44f8130b7e1144fef4554fd972b9df28d70bdfee2b20f5c7c32c1ab7274 - Sigstore transparency entry: 937226548
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp312-cp312-win_amd64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp312-cp312-win_amd64.whl
- Upload date:
- Size: 13.6 MB
- Tags: CPython 3.12, Windows x86-64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
3f102278abd4dcc2ced53de5ec678cdb7d0b991419ca1d16b29b2aa512968163
|
|
| MD5 |
5f9e6a65939a8a04c31b0b1bfc8dec37
|
|
| BLAKE2b-256 |
ea85ee4a25eaeab7ac875329da5be8ac40437a2f596b62bc1f9b67962513d890
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp312-cp312-win_amd64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp312-cp312-win_amd64.whl -
Subject digest:
3f102278abd4dcc2ced53de5ec678cdb7d0b991419ca1d16b29b2aa512968163 - Sigstore transparency entry: 937226596
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp312-cp312-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp312-cp312-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl
- Upload date:
- Size: 15.5 MB
- Tags: CPython 3.12, manylinux: glibc 2.17+ x86-64, manylinux: glibc 2.5+ x86-64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d17fde7f4484579251f194f4260e98b8839b3e24fb7713082d8977dab7b11bdc
|
|
| MD5 |
017502ab9971dcaaf5f29c3373206af6
|
|
| BLAKE2b-256 |
62bd8c23cf3fa6e2a1011a8b3f8094aaecad4abb42ff1a6a9cda1416e51f0caf
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp312-cp312-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp312-cp312-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl -
Subject digest:
d17fde7f4484579251f194f4260e98b8839b3e24fb7713082d8977dab7b11bdc - Sigstore transparency entry: 937226631
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp312-cp312-macosx_11_0_arm64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp312-cp312-macosx_11_0_arm64.whl
- Upload date:
- Size: 14.8 MB
- Tags: CPython 3.12, macOS 11.0+ ARM64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d2058f6dbbcd7e95a6d4d0b9323855f56f630ccce26260eac17c24a9673885b8
|
|
| MD5 |
1cb29b717625139bdfa3a52abaed069b
|
|
| BLAKE2b-256 |
fd2b64727dfe1414ae5b28ca00daca3d24e1b496e2898a9d130934930589b7c9
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp312-cp312-macosx_11_0_arm64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp312-cp312-macosx_11_0_arm64.whl -
Subject digest:
d2058f6dbbcd7e95a6d4d0b9323855f56f630ccce26260eac17c24a9673885b8 - Sigstore transparency entry: 937226518
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp312-cp312-macosx_10_13_x86_64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp312-cp312-macosx_10_13_x86_64.whl
- Upload date:
- Size: 13.9 MB
- Tags: CPython 3.12, macOS 10.13+ x86-64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
7ee435c546ef4f438755b3b3f0c0c5f26a88cf87ee0e86693da1aad6b53b9890
|
|
| MD5 |
68351c3b02e5c131ceffa1099f81b89c
|
|
| BLAKE2b-256 |
8f27f79d5f190fa289fb75db251f6d888ead7af9079e69df764049daddd9c9dc
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp312-cp312-macosx_10_13_x86_64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp312-cp312-macosx_10_13_x86_64.whl -
Subject digest:
7ee435c546ef4f438755b3b3f0c0c5f26a88cf87ee0e86693da1aad6b53b9890 - Sigstore transparency entry: 937226557
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp311-cp311-win_amd64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp311-cp311-win_amd64.whl
- Upload date:
- Size: 13.6 MB
- Tags: CPython 3.11, Windows x86-64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d62f52ee220998e3222a8e3ff8182c13f83684b087b138e2a583715647c32d3d
|
|
| MD5 |
315e07ce5231b1ae05176457fa5d62d4
|
|
| BLAKE2b-256 |
a6fe0143b12ab741d5c4b92d40b06261cdf87f4b0eb82023aa99532f35fb46ab
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp311-cp311-win_amd64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp311-cp311-win_amd64.whl -
Subject digest:
d62f52ee220998e3222a8e3ff8182c13f83684b087b138e2a583715647c32d3d - Sigstore transparency entry: 937226580
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp311-cp311-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp311-cp311-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl
- Upload date:
- Size: 15.5 MB
- Tags: CPython 3.11, manylinux: glibc 2.17+ x86-64, manylinux: glibc 2.5+ x86-64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
3d1066337619a66f38e0cc2cc23d24e23f4348e682271054cc9a73d37f8e921f
|
|
| MD5 |
099160e0b102397708f4fba46cd15d12
|
|
| BLAKE2b-256 |
9ceff50d29874baa59b4c1d6b37bf4f5a65fca2469e8cd2ac1ebe4a454781bd4
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp311-cp311-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp311-cp311-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl -
Subject digest:
3d1066337619a66f38e0cc2cc23d24e23f4348e682271054cc9a73d37f8e921f - Sigstore transparency entry: 937226508
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp311-cp311-macosx_11_0_arm64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp311-cp311-macosx_11_0_arm64.whl
- Upload date:
- Size: 14.8 MB
- Tags: CPython 3.11, macOS 11.0+ ARM64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
72a848c8ea237565601727ce398ecef457b17e27f976d962c1a5bc1f72d6c6c8
|
|
| MD5 |
cb00ce0787b9fa76872db96317de39cf
|
|
| BLAKE2b-256 |
9d66291297259ca01891befecf115b24b9e304740f12d3733d4cb3f1af86db47
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp311-cp311-macosx_11_0_arm64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp311-cp311-macosx_11_0_arm64.whl -
Subject digest:
72a848c8ea237565601727ce398ecef457b17e27f976d962c1a5bc1f72d6c6c8 - Sigstore transparency entry: 937226603
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp311-cp311-macosx_10_9_x86_64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp311-cp311-macosx_10_9_x86_64.whl
- Upload date:
- Size: 13.9 MB
- Tags: CPython 3.11, macOS 10.9+ x86-64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
425ffbf2bf1563fedc07232413f21f36681c91ca89c5ada29eebfe14dc3ad722
|
|
| MD5 |
a2bfe38c5694bcb3a332dcd8dd2a23f1
|
|
| BLAKE2b-256 |
7f410108a6442472545d3f8136ff78981c53a74ae7caa305bc52c3711003edcd
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp311-cp311-macosx_10_9_x86_64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp311-cp311-macosx_10_9_x86_64.whl -
Subject digest:
425ffbf2bf1563fedc07232413f21f36681c91ca89c5ada29eebfe14dc3ad722 - Sigstore transparency entry: 937226656
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp310-cp310-win_amd64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp310-cp310-win_amd64.whl
- Upload date:
- Size: 13.6 MB
- Tags: CPython 3.10, Windows x86-64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
6b2ac4da06264db8a1ea07c0b98e4ecf66b1ae893b0ddd01b79694b6ca0459fb
|
|
| MD5 |
f58c4208d3071211f2c4be69e24fd81a
|
|
| BLAKE2b-256 |
5eaf0b6190a3818cc1ac9a6d31086e171c8af47fbfdce1fc898c14a4fc521399
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp310-cp310-win_amd64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp310-cp310-win_amd64.whl -
Subject digest:
6b2ac4da06264db8a1ea07c0b98e4ecf66b1ae893b0ddd01b79694b6ca0459fb - Sigstore transparency entry: 937226615
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp310-cp310-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp310-cp310-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl
- Upload date:
- Size: 15.5 MB
- Tags: CPython 3.10, manylinux: glibc 2.17+ x86-64, manylinux: glibc 2.5+ x86-64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
cb965bfeb0961b10ece03d35c3a71c79b1342e2a62d5a46ef0237ecdfa25539d
|
|
| MD5 |
06cfefdf4018b134af7e0cd82907ba26
|
|
| BLAKE2b-256 |
a72ada8d0c5bb062ecf855ac90d5797846ed95b824b4ab9670dccd75a9754db9
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp310-cp310-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp310-cp310-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl -
Subject digest:
cb965bfeb0961b10ece03d35c3a71c79b1342e2a62d5a46ef0237ecdfa25539d - Sigstore transparency entry: 937226672
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp310-cp310-macosx_11_0_arm64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp310-cp310-macosx_11_0_arm64.whl
- Upload date:
- Size: 14.8 MB
- Tags: CPython 3.10, macOS 11.0+ ARM64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
4e7d159301c86272b4a14ddd650cfb30512a8afabb68d3dbafc7b1f53c74f69e
|
|
| MD5 |
aba47b9064a009d6dd3ed3753a887f38
|
|
| BLAKE2b-256 |
cfe3945b99fc435ef99ae5e8c56fa765d89e797f50189e8f7eba97506c98c9b5
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp310-cp310-macosx_11_0_arm64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp310-cp310-macosx_11_0_arm64.whl -
Subject digest:
4e7d159301c86272b4a14ddd650cfb30512a8afabb68d3dbafc7b1f53c74f69e - Sigstore transparency entry: 937226539
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp310-cp310-macosx_10_9_x86_64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp310-cp310-macosx_10_9_x86_64.whl
- Upload date:
- Size: 13.9 MB
- Tags: CPython 3.10, macOS 10.9+ x86-64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
7a6435573347fbc5c7debb95f7b1f2f34ce66058b3b8351a257fe96b564ed101
|
|
| MD5 |
92bbbaffb06f2d4ccfad22f04fa263f8
|
|
| BLAKE2b-256 |
70f1125b3a3a6ceb8d4ad15ad72c78152c6397f1e22d95cb492bce13e0df0c8c
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp310-cp310-macosx_10_9_x86_64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp310-cp310-macosx_10_9_x86_64.whl -
Subject digest:
7a6435573347fbc5c7debb95f7b1f2f34ce66058b3b8351a257fe96b564ed101 - Sigstore transparency entry: 937226531
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp39-cp39-win_amd64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp39-cp39-win_amd64.whl
- Upload date:
- Size: 13.6 MB
- Tags: CPython 3.9, Windows x86-64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
8d6bdbdf963cd5f5e05e44645190cb17b226658e02c13964b3685d78a466f0c3
|
|
| MD5 |
4f96db160988db770712e7f2da682166
|
|
| BLAKE2b-256 |
46948225d8cfdb51f395bfbae7d131275590cedad596e4e63e0782ef9c0a5ee9
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp39-cp39-win_amd64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp39-cp39-win_amd64.whl -
Subject digest:
8d6bdbdf963cd5f5e05e44645190cb17b226658e02c13964b3685d78a466f0c3 - Sigstore transparency entry: 937226701
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp39-cp39-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp39-cp39-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl
- Upload date:
- Size: 15.5 MB
- Tags: CPython 3.9, manylinux: glibc 2.17+ x86-64, manylinux: glibc 2.5+ x86-64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
23c5ad6a576ba0c9ffba48225882c3dacb2200d08ef12e060d4767988ca0f6c0
|
|
| MD5 |
39442f8c2b0187fa95a15632bccbac51
|
|
| BLAKE2b-256 |
872cdccc49f62b882174bc825fb2365e5e50418edfb5c13e61c301a85cc355b6
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp39-cp39-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp39-cp39-manylinux_2_5_x86_64.manylinux1_x86_64.manylinux_2_17_x86_64.manylinux2014_x86_64.whl -
Subject digest:
23c5ad6a576ba0c9ffba48225882c3dacb2200d08ef12e060d4767988ca0f6c0 - Sigstore transparency entry: 937226587
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp39-cp39-macosx_11_0_arm64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp39-cp39-macosx_11_0_arm64.whl
- Upload date:
- Size: 14.8 MB
- Tags: CPython 3.9, macOS 11.0+ ARM64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
4c5439347b9e173c06e6803fe8542d2296dd0cea27f744144ce21c949ce70266
|
|
| MD5 |
564c9d0a7214b3de13a0c1f2723b6417
|
|
| BLAKE2b-256 |
45f6313f1dd5d7b9aa4637ad58bfc6879b204c5c7667bbb078cb09d32a5a97da
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp39-cp39-macosx_11_0_arm64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp39-cp39-macosx_11_0_arm64.whl -
Subject digest:
4c5439347b9e173c06e6803fe8542d2296dd0cea27f744144ce21c949ce70266 - Sigstore transparency entry: 937226685
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:
File details
Details for the file ms2pip-4.1.2-cp39-cp39-macosx_10_9_x86_64.whl.
File metadata
- Download URL: ms2pip-4.1.2-cp39-cp39-macosx_10_9_x86_64.whl
- Upload date:
- Size: 13.9 MB
- Tags: CPython 3.9, macOS 10.9+ x86-64
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
46006b08e9f44f20224a7435741996f590e18b626dfb05d475714083d809e54e
|
|
| MD5 |
c4691c123a2bcd976fe63ebafb335d0b
|
|
| BLAKE2b-256 |
006b27d68f0d1642646e597cc4390f7710a1675b9115ed1e0140da778a05e022
|
Provenance
The following attestation bundles were made for ms2pip-4.1.2-cp39-cp39-macosx_10_9_x86_64.whl:
Publisher:
build_and_publish.yml on CompOmics/ms2pip
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
ms2pip-4.1.2-cp39-cp39-macosx_10_9_x86_64.whl -
Subject digest:
46006b08e9f44f20224a7435741996f590e18b626dfb05d475714083d809e54e - Sigstore transparency entry: 937226697
- Sigstore integration time:
-
Permalink:
CompOmics/ms2pip@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Branch / Tag:
refs/tags/v4.1.2 - Owner: https://github.com/CompOmics
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
build_and_publish.yml@0aca6b32b86f34a0c7bbb116056a6a43676796aa -
Trigger Event:
release
-
Statement type:







