A multi-sample fastqc pipeline from Sequana project

Project description




This is is the fastqc pipeline from the Sequana projet
- Overview:
Runs fastqc and multiqc on a set of Sequencing data to produce control quality reports
- Input:
A set of FastQ files (paired or single-end) compressed or not
- Output:
An HTML file summary.html (individual fastqc reports, mutli-samples report)
- Status:
Production
- Wiki:
- Documentation:
This README file, the Wiki from the github repository (link above) and https://sequana.readthedocs.io
- Citation:
Cokelaer et al, (2017), ‘Sequana’: a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI https://doi:10.21105/joss.00352
Installation
sequana_fastqc is based on Python3, just install the package as follows:
pip install sequana_fastqc --upgrade
You will need third-party software such as fastqc. Please see below for details.
Usage
If you have a set of FastQ files in a data/ directory, type:
sequana_fastqc --input-directory data
To know more about the options (e.g., add a different pattern to restrict the execution to a subset of the input files, change the output/working directory, etc):
sequana_fastqc --help
The call to sequana_fastqc creates a directory fastqc. Then, you go to the working directory and execute the pipeline as follows:
cd fastqc sh fastqc.sh # for a local run
This launch a snakemake pipeline. If you are familiar with snakemake, you can retrieve the fastqc.rules and config.yaml files and then execute the pipeline yourself with specific parameters:
snakemake -s fastqc.rules --cores 4 --stats stats.txt
Or use sequanix interface.
Please see the Wiki for more examples and features.
Tutorial
You can retrieve test data from sequana_fastqc (https://github.com/sequana/fastqc) or type:
wget https://raw.githubusercontent.com/sequana/fastqc/master/sequana_pipelines/fastqc/data/data_R1_001.fastq.gz wget https://raw.githubusercontent.com/sequana/fastqc/master/sequana_pipelines/fastqc/data/data_R2_001.fastq.gz
then, prepare the pipeline:
sequana_fastqc --input-directory . cd fastqc sh fastq.sh # once done, remove temporary files (snakemake and others) make clean
Just open the HTML entry called summary.html. A multiqc report is also available. You will get expected images such as the following one:
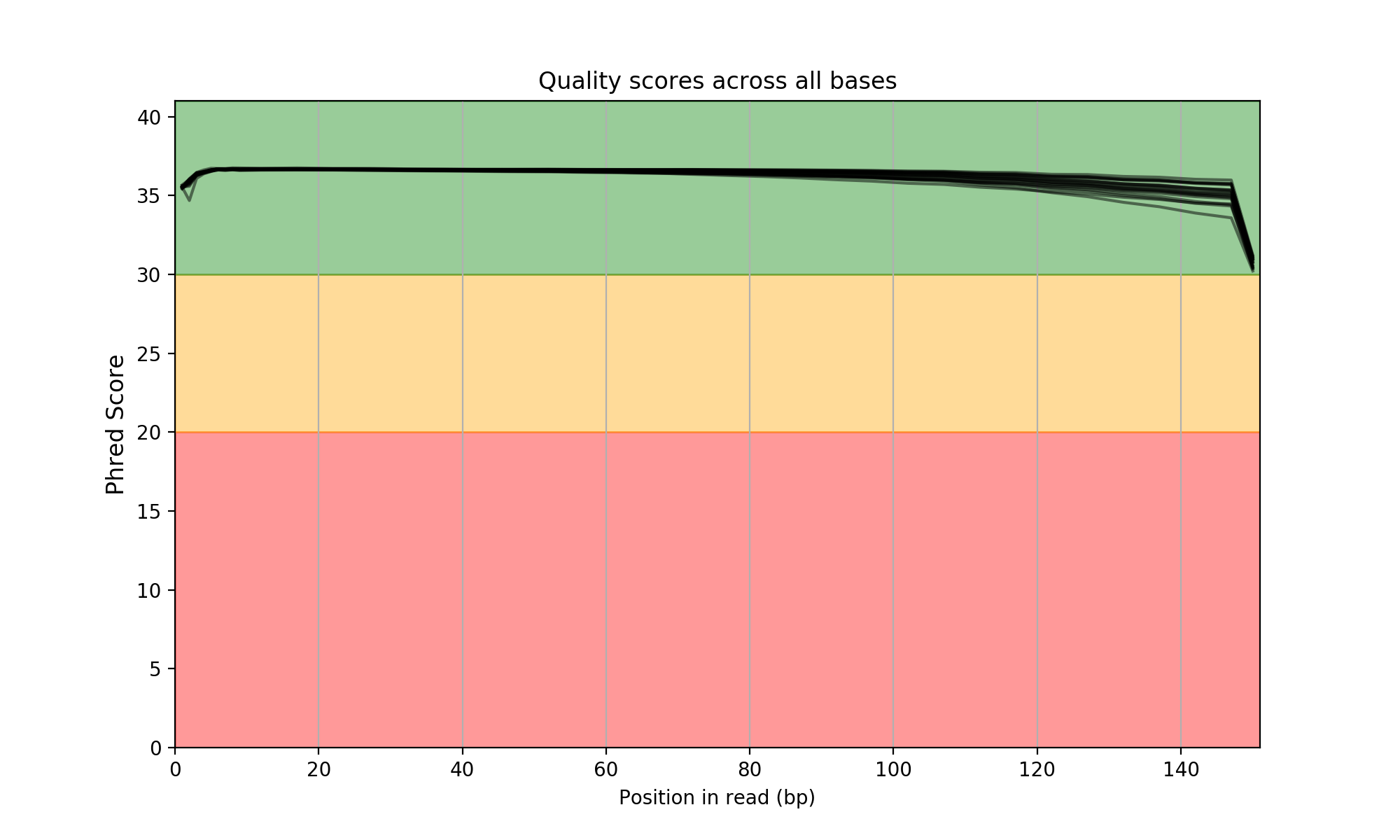
Please see the Wiki for more examples and features.
Requirements
This pipelines requires the following executable(s):
fastqc
falco (optional)
For Linux users, we provide apptainer/singularity images available through the damona project (https://damona.readthedocs.io).
To make use of them, initiliase the pipeline with the –use-apptainer option and everything should be downloaded automatically for you, which also guarantees reproducibility:
sequana_fastqc --input-directory data --use-apptainer --apptainer-prefix ~/images
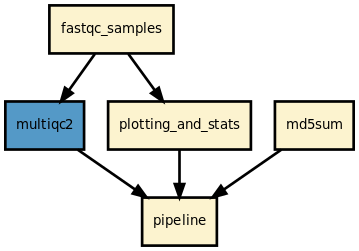
Details
This pipeline runs fastqc in parallel on the input fastq files (paired or not) and then execute multiqc. A brief sequana summary report is also produced. s You may use falco instead of fastqc. This is experimental but seem to work for Illumina/FastQ files.
This pipeline has been tested on several hundreds of MiSeq, NextSeq, MiniSeq, ISeq100, Pacbio runs.
It produces a md5sum of your data. It copes with empty samples. Produces ready-to-use HTML reports, etc
Rules and configuration details
Here is the latest documented configuration file to be used with the pipeline. Each rule used in the pipeline may have a section in the configuration file.
Changelog
Version |
Description |
|---|---|
1.8.2 |
|
1.8.1 |
|
1.8.0 |
|
1.7.1 |
|
1.7.0 |
|
1.6.2 |
|
1.6.1 |
|
1.6.0 |
|
1.5.0 |
|
1.4.2 |
|
1.4.1 |
|
1.4.0 |
|
1.3.0 |
|
1.2.0 |
|
1.1.0 |
|
1.0.1 |
|
1.0.0 |
|
0.9.15 |
|
0.9.14 |
|
0.9.13 |
|
0.9.12 |
|
0.9.11 |
|
0.9.10 |
|
0.9.9 |
|
0.9.8 |
|
0.9.7 |
|
0.9.6 |
add the readtag option |
Contribute & Code of Conduct
To contribute to this project, please take a look at the Contributing Guidelines first. Please note that this project is released with a Code of Conduct. By contributing to this project, you agree to abide by its terms.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file sequana_fastqc-1.9.0.tar.gz.
File metadata
- Download URL: sequana_fastqc-1.9.0.tar.gz
- Upload date:
- Size: 91.5 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/5.1.1 CPython/3.12.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
5deb9aff418262a50650df3b5f90e9b56e8bb9cb4b7e730d095fbfef2a371934
|
|
| MD5 |
f398e81f11df44768739c949db08f916
|
|
| BLAKE2b-256 |
13b7a5a25b1edeae33392aca798bb800038a9fd520c13224b26850f38fff0050
|
File details
Details for the file sequana_fastqc-1.9.0-py3-none-any.whl.
File metadata
- Download URL: sequana_fastqc-1.9.0-py3-none-any.whl
- Upload date:
- Size: 89.1 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/5.1.1 CPython/3.12.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
ee6e0febb11630d54eaa12afaae94fc78cc871a3fa5b245576d739a0852f63b5
|
|
| MD5 |
e3431352ed6016360a5642de11c83b89
|
|
| BLAKE2b-256 |
5130ef207570cd07f70fdff2d7a6e09eb6bcf96c0302e3e1418d04b532d922dc
|







