Converter between hic files (from juicer) and single-resolution or multi-resolution cool files (for cooler). Both hic and cool files describe Hi-C contact matrices. Intended to be lightweight, this can be used as an imported package or a stand-alone Python tool for command line conversion.




Project description
hic2cool

Converter between hic files (from juicer) and single-resolution or multi-resolution cool files (for cooler). Both hic and cool files describe Hi-C contact matrices. Intended to be lightweight, this can be used as an imported package or a stand-alone Python tool for command line conversion
The hic parsing code is based off the straw project by Neva C. Durand and Yue Wu. The hdf5-based structure used for cooler file writing is based off code from the cooler repository.
Important
-
Starting from version 0.8.0, hic2cool no longer supports Python 2.7.
-
If you converted a hic file using a version of hic2cool lower than 0.5.0, please update your cooler file with the new update function.
Using the Python package
$ pip install hic2cool
You can also download the code directly and install using Poetry (as of version 1.0.0)
$ poetry install
Once the package is installed, the main method is hic2cool_convert. It takes the same parameters as hic2cool.py, described in the next section. Example usage in a Python script is shown below or in test.py.
from hic2cool import hic2cool_convert
hic2cool_convert(<infile>, <outfile>, <resolution (optional)>, <nproc (optional)>, <warnings (optional)>, <silent (optional)>)
Converting files using the command line
The main use of hic2cool is converting between filetypes using hic2cool convert. If you install hic2cool itself using pip, you use it on the command line with:
$ hic2cool convert <infile> <outfile> -r <resolution> -p <nproc>
Arguments for hic2cool convert
infile is a .hic input file.
outfile is a .cool output file.
-r, or --resolution, is an integer bp resolution supported by the hic file. Please note that only resolutions contained within the original hic file can be used. If 0 is given, will use all resolutions to build a multi-resolution file. Default is 0.
-p, or --nproc, is the number of processes to use. Default 1. The multiprocessing is not very efficient and would slightly improve speed only for large high-resolution matrices.
-w, or --warnings, causes warnings to be explicitly printed to the console. This is false by default, though there are a few cases in which hic2cool will exit with an error based on the input hic file.
-s, or --silent, run in silent mode and hide console output from the program. Default false.
-v, or --version, print out hic2cool package version and exit.
-h, or --help, print out help about the package/specific run mode and exit.
Running hic2cool from the command line will cause some helpful information about the hic file to be printed to stdout unless the -s flag is used.
Output file structure
If you elect to use all resolutions, a multi-resolution .mcool file will be produced. This changes the hdf5 structure of the file from a typical .cool file. Namely, all of the information needed for a complete cooler file is stored in separate hdf5 groups named by the individual resolutions. The hdf5 hierarchy is organized as such:
File --> 'resolutions' --> '###' (where ### is the resolution in bp). For example, see the code below that generates a multi-res file and then accesses the specific resolution of 10000 bp.
from hic2cool import hic2cool_convert
import cooler
### using 0 triggers a multi-res output
hic2cool_convert('my_hic.hic', 'my_cool.cool', 0)
### will give you the cooler object with resolution = 10000 bp
my_cooler = cooler.Cooler('my_cool.cool::resolutions/10000')
When using only one resolution, the .cool file produced stores all the necessary information at the top level. Thus, organization in the multi-res format is not needed. The code below produces a file with one resolution, 10000 bp, and opens it with a cooler object.
from hic2cool import hic2cool_convert
import cooler
### giving a specific resolution below (e.g. 10000) triggers a single-res output
hic2cool_convert('my_hic.hic', 'my_cool.cool', 10000)
h5file = h5py.File('my_cool.cool', 'r')
### will give you the cooler object with resolution = 10000 bp
my_cooler = cooler.Cooler(h5file)
higlass
Multi-resolution coolers produced by hi2cool can be visualized using higlass. Please note that single resolution coolers are NOT higlass compatible (created when using a non-zero value for -r). If you created a cooler before hic2cool version 0.5.0 that you want to view in higlass, it is highly recommended that you upgrade it before viewing on higlass to ensure correct normalization behavior.
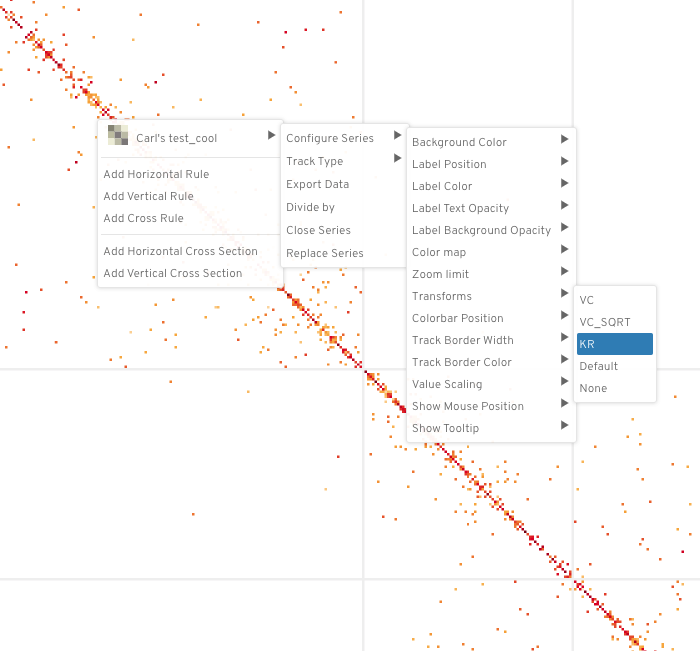
To apply the hic normalization transformations in higlass, right click on the tileset and do the following:
"<name of tileset>" --> "Configure Series" --> "Transforms" --> "<norm>"
Updating hic2cool coolers
As of hic2cool version 0.5.0, there was a critical change in how hic normalization vectors are handled in the resulting cooler files. Prior to 0.5.0, hic normalization vectors were inverted by hic2cool. The rationale for doing this is that hic uses divisive normalization values, whereas cooler uses multiplicative values. However, higlass and the 4DN analysis pipelines specifically handled the divisive normalization values, so hic2cool now handles them the same way.
In the near future, there will be a cooler package release to correctly handle divisive hic normalization values when balancing.
To update a hic2cool cooler, simply run:
hic2cool update <infile> <outfile (optional)>
If you only provide the infile argument, then the cooler will be updated directly. If you provide an optional outfile file path, then a new cooler updated cooler file will be created and the original file will remain unchanged.
Extracting hic normalization values
As of hic2cool 0.5.0, you can easily extract hic normalization vectors to an existing cooler file. This will only work if the specified cooler file shares the resolutions found in the hic file. To do this, simply run:
hic2cool extract-norms <hic file> <cooler file>
You may also provide the optional -e flag, which will cause the mitchondrial chromosome to automatically be omitted from the extraction. This is found by name; the code specifically looks for one of ['M', 'MT', 'chrM', 'chrMT'] (in a case-insensitive way). Just like with hic2cool convert, you can also provide -s and -w arguments.
Changelog
1.0.1
- Restore command line usage, adds missing README update
1.0.0
- Switch to poetry, upgraded
python,numpy,coolerversions, h5file I/O clean up, replacemultiprocessingwith `multiprocess'
0.8.3
- Partial fix for zlib decompression issue.
0.8.2
- loosened version for
numpy,scipyandpandas.
0.8.1
setup.pytakes dependencies directly fromrequirements.txt(requirements.txtupdated to matchsetup.py)
0.8.0
- multiprocessing support for convert
- change in usage of convert API due to the addition of the
nprocoption - Python 2.7 is deprecated.
0.7.3
- Pinned
pandas==0.24.2since newer versions deprecate python 2
0.7.2
- Warning from
hic2cool_utils.parse_hicwill now output chromsome names, not indices
0.7.1
- Add
formatandformat-versionto/collection for multi-resolution coolers written by hic2cool - Run
hic2cool_updateto add these attributes to mcool files generated with previous hic2cool versions - Fixed issue where datetime-derived metadata was written as bytestring when using python 2
0.7.0
- Fixed package issues associated with python 2
- Fixed issue where some cooler metadata was written as non-unicode when using python 2
0.6.1
- Fixed input issue with
hic2cool updatewhen using python 2
0.6.0
- Added
format-versionandstorage-typeto attributes of output cooler to get up-to-date with cooler schema v3 - Run
hic2cool updateto add these attributes to files generated with previous hic2cool versions
0.5.1
Fixed packaging issue by adding MANIFEST.in and made some documentation/pypi edits
0.5.0
Large release that changes how hic2cool is run
- hic2cool is now executed with
hic2cool <mode>, where mode is one of:[convert, update, extract-norms] - Added two new modes:
update(update coolers made by hic2cool based on version) andextract-norms(extract hic normalization vectors to pre-existing cooler file) - Removed old hic2cool_extractnorms script (this is now run with
hic2cool extract-norms) - hic normalization vectors are NO LONGER INVERTED when added to output cooler for consistency with the 4DN omics processing pipeline and higlass
- Missing hic normalization vectors are now represented by vectors of
NaN(used to be vectors of zeros) - Improvement of help messages when running hic2cool and change around arguments for running the program
- Test updates
0.4.2
- Fixed issue where hic files could not be converted if they were missing normalization vectors
0.4.1
- Fixed error in reading counts from hic files of version 6
- Chromosome names are now directly taken from hic file (with exception of 'all')
0.4.0
Large patch, should fix most memory issues and improve runtimes:
- Changed run parameters. Removed -n and -e; added -v (--version) and -w (--warnings)
- Improved memory usage
- Improved runtime (many thanks to Nezar Abdennur)
- hic2cool now does a 'direct' conversion of files and does not fail on missing chr-chr contacts or missing normalization vectors. Finding these issues will cause warnings to be printed (controlled by -w flag)
- No longer uses the 'weights' column, which is reserved for cooler
- No longer takes a normalization type argument. All normalization vectors from the hic file are automatically added to the bins table in the output .cool
- Many other minor bug fixes/code improvement
0.3.7
Fixed issue with bin1_offset not containing final entry (should be length nbins + 1).
0.3.6
Simple release to fix pip execution.
0.3.5
README updates, switched cooler syntax in test, and added helpful printing of hic file header info when using the command line tool.
0.3.4
Fixed issue where chromosome name was not getting properly set for 'All' vs 'all'.
0.3.3
Removed rounding fix. For now, allow py2 and py3 weights to have different number of significant figures (they're very close).
0.3.2
Changed output file structure for single resolution file. Resolved an issue where rounding for weights was different between python 2 and 3.
0.3.1
Added .travis.yml for automated testing. Changed command line running scheme. Python3 fix in hic2cool_utils.
0.3.0
Added multi-resolution format to output cool files. Setup argparse. Improved speed. Added tests for new resolutions format.
Contributors
Written by Carl Vitzthum (1), Nezar Abdennur (2), Soo Lee (1), and Peter Kerpedjiev (3).
(1) Park lab, Harvard Medical School DBMI
(2) Mirny lab, MIT
(3) Gehlenborg lab, Harvard Medical School DBMI
Originally published 1/26/17.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file hic2cool-1.0.1.tar.gz.
File metadata
- Download URL: hic2cool-1.0.1.tar.gz
- Upload date:
- Size: 25.1 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: poetry/1.6.1 CPython/3.8.13 Darwin/22.6.0
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d1c9ffe5d3e03027774ac0abab7c0102704954ff8cf32d839d385ada727c3633
|
|
| MD5 |
ac67c4d74871cb2dcd782f7df2c7a740
|
|
| BLAKE2b-256 |
1c31da0499e70be579ff206980ee49fb60a1cd1d5acc3739a10c3be359d9def3
|
File details
Details for the file hic2cool-1.0.1-py3-none-any.whl.
File metadata
- Download URL: hic2cool-1.0.1-py3-none-any.whl
- Upload date:
- Size: 23.6 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: poetry/1.6.1 CPython/3.8.13 Darwin/22.6.0
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
c8823d2e47554d4b0465ce4b699297b2a628ff22b2f6a33ef0b668dd60fb7397
|
|
| MD5 |
5919a3dd832bcb6a6081048dac4ba137
|
|
| BLAKE2b-256 |
f858bf9706a891f857b6f3bfcd6d1f3b490d0182dda29406e8c8267c69413e23
|







